As doenças linfoproliferativas constituem um grupo heterogêneo de neoplasias que afetam o sistema linfático. Uma distinção fundamental neste grupo é feita entre o Linfoma de Hodgkin (LH) e os Linfomas Não Hodgkin (LNH). O LH, foco deste artigo, é definido pela presença característica das células de Reed-Sternberg (CRS), distinguindo-se histologicamente dos LNH.
Introdução ao Linfoma de Hodgkin (LH): Definição, Epidemiologia e Etiopatogenia
O Linfoma de Hodgkin, também denominado Doença de Hodgkin, é uma neoplasia maligna que se origina nos linfócitos B, especificamente nos centros germinativos ou pós-germinativos dos tecidos linfoides, mais comumente nos linfonodos. Representa uma entidade distinta dentro dos linfomas, caracterizada por aspectos epidemiológicos, etiopatogênicos e histopatológicos particulares.
Definição e Características Gerais
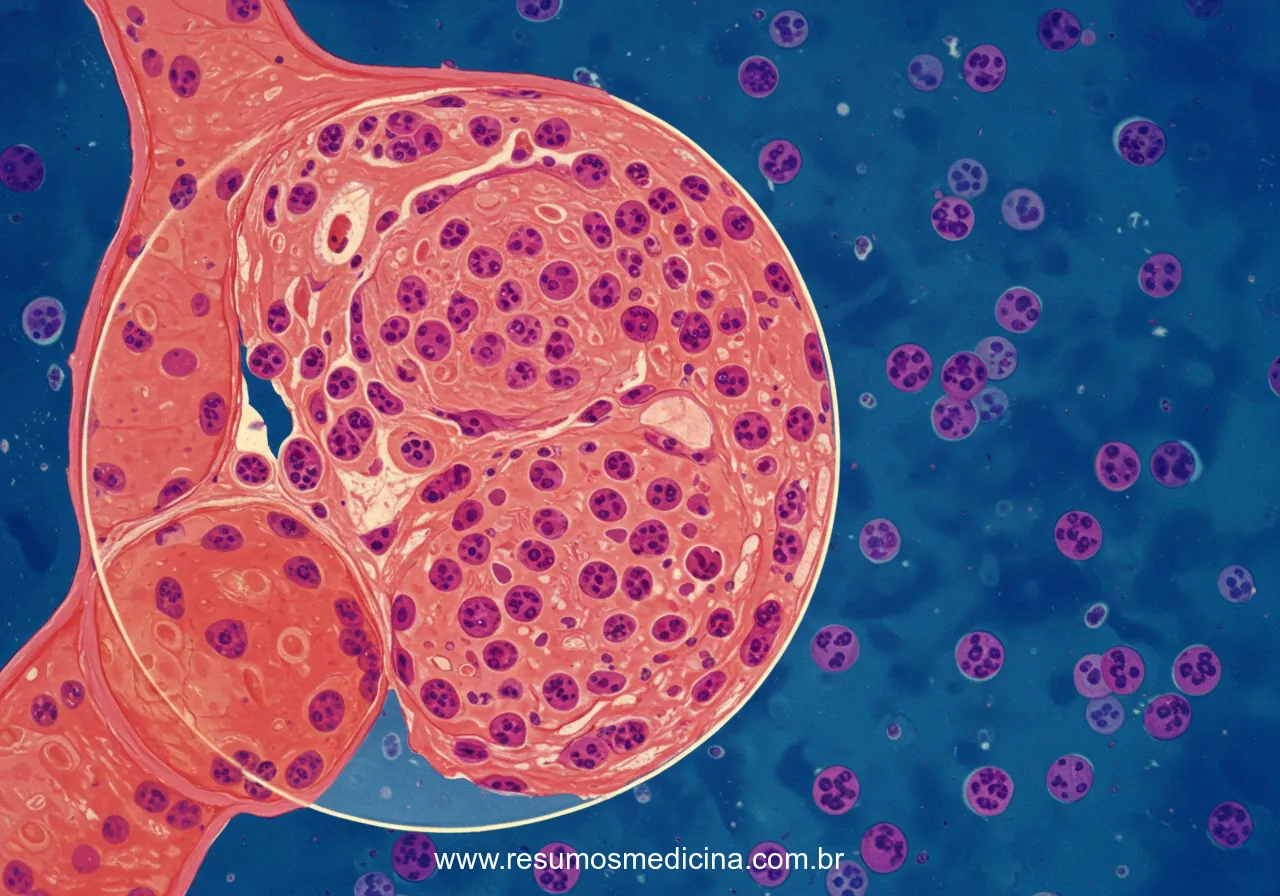
O LH é definido pela presença das células de Reed-Sternberg (CRS) e suas variantes morfológicas. Estas são células neoplásicas grandes, frequentemente multinucleadas ou bilobadas, com nucléolos proeminentes (aspecto de ‘olhos de coruja’), derivadas de linfócitos B. Uma característica marcante do LH é que as CRS constituem uma minoria da massa tumoral (frequentemente < 2%), estando imersas em um extenso e heterogêneo microambiente inflamatório reativo, composto por linfócitos T e B não neoplásicos, eosinófilos, neutrófilos, plasmócitos, histiócitos e fibroblastos.
Epidemiologia
A incidência do Linfoma de Hodgkin exibe um padrão bimodal característico na distribuição etária, com um primeiro pico em adultos jovens (faixa etária de 15-35 anos) e um segundo pico em indivíduos com idade mais avançada (geralmente acima de 55 anos). Embora seja menos prevalente que o conjunto dos LNH, o LH constitui uma fração importante das malignidades linfoides diagnosticadas globalmente.
Etiopatogenia
A etiologia do LH é multifatorial e não completamente compreendida, envolvendo a interação de fatores genéticos, imunológicos e ambientais. Fatores de risco estabelecidos incluem predisposição genética sugerida por casos familiares, estados de imunodeficiência (congênita ou adquirida, como infecção pelo HIV ou imunossupressão pós-transplante) e a infecção prévia pelo vírus Epstein-Barr (EBV). A associação com o EBV é particularmente observada em certos subtipos histológicos e populações específicas.
Fisiopatologia
Acredita-se que a patogênese do LH envolva a transformação maligna de linfócitos B do centro germinativo que, por mecanismos aberrantes, escaparam dos processos normais de apoptose. As células de Reed-Sternberg resultantes, embora neoplásicas, não são eficientes na apresentação de antígenos e sobrevivem através da manipulação do microambiente. Elas secretam um perfil complexo de citocinas e quimiocinas, responsáveis pelo recrutamento e ativação das diversas células inflamatórias que compõem a maior parte da massa tumoral. Esta interação dinâmica entre as CRS e o infiltrado reativo é fundamental não apenas para a sobrevivência e proliferação das células neoplásicas, mas também contribui significativamente para as manifestações clínicas locais e sistêmicas da doença, como os sintomas B. Diferente de muitos LNH, o LH tende a disseminar-se de forma contígua através das cadeias linfonodais adjacentes. Clinicamente, o LH é considerado uma neoplasia com bom prognóstico geral, apresentando altas taxas de cura, especialmente quando diagnosticado e tratado em estágios iniciais.
Manifestações Clínicas: Sintomas e Apresentação Comum
A apresentação clínica mais frequente do Linfoma de Hodgkin (LH) é a linfadenopatia indolor, que tipicamente acomete as cadeias ganglionares cervicais, supraclaviculares e mediastinais, embora linfonodos axilares e inguinais também possam ser afetados. Uma característica importante do LH é a sua tendência à disseminação por contiguidade, ou seja, a progressão ordenada para grupos linfonodais adjacentes, um padrão que o distingue frequentemente dos linfomas não Hodgkin (LNH).
Sintomas Sistêmicos (Sintomas B)
Aproximadamente um terço dos pacientes apresenta sintomas sistêmicos, conhecidos como ‘Sintomas B’, que possuem relevância prognóstica e influenciam o estadiamento da doença. A presença destes sintomas indica, geralmente, uma doença mais ativa e está associada a um prognóstico menos favorável. Eles são definidos como:
- Febre: Temperatura inexplicada superior a 38°C, persistente ou recorrente.
- Sudorese Noturna: Episódios de transpiração profusa durante a noite, que podem requerer a troca de roupas de cama.
- Perda de Peso Inexplicada: Perda involuntária de mais de 10% do peso corporal nos seis meses anteriores ao diagnóstico.
Estes sintomas constitucionais, assim como o prurido, são atribuídos em parte à liberação de citocinas pelas células de Reed-Sternberg e pelo microambiente inflamatório reativo.
Outras Manifestações Clínicas
- Prurido: O prurido generalizado, por vezes intenso, é um sintoma relativamente comum no LH, embora não seja classificado como um sintoma B.
- Fadiga: Sensação de cansaço ou exaustão pode ser relatada pelos pacientes.
- Dor Induzida por Álcool: Um sintoma clássico, embora raro, é a dor nos linfonodos acometidos logo após a ingestão de bebidas alcoólicas (‘dor de Hodgkin’). O mecanismo fisiopatológico subjacente a este fenômeno não é totalmente compreendido, mas pode envolver vasodilatação ou liberação de mediadores inflamatórios nos linfonodos neoplásicos.
- Envolvimento Mediastinal: O acometimento de linfonodos mediastinais é frequente, especialmente no subtipo Esclerose Nodular. Massas mediastinais volumosas podem causar sintomas compressivos, como tosse seca persistente, dispneia, dor torácica ou síndrome da veia cava superior.
- Esplenomegalia: O envolvimento do baço pode levar ao seu aumento (esplenomegalia), detectável ao exame físico ou por imagem.
- Acometimento Extranodal: O envolvimento primário ou secundário de órgãos fora do sistema linfático é menos comum no LH do que nos LNH, ocorrendo geralmente em fases mais avançadas (Estádio IV). Os sítios extranodais mais frequentemente afetados incluem fígado, pulmões e medula óssea. O envolvimento por extensão direta de um sítio nodal adjacente é classificado como ‘E’ no sistema de estadiamento.
- Alterações Hematológicas: Podem ocorrer diversas manifestações hematológicas, incluindo anemia (normocítica ou, menos frequentemente, autoimune hemolítica), trombocitopenia ou trombocitose, e leucopenia ou leucocitose (por vezes com eosinofilia). Estas alterações podem ser resultado da infiltração medular, de fenômenos autoimunes ou paraneoplásicos, ou efeitos adversos do tratamento.
Sintomas B: Definição, Significado Clínico e Prognóstico no Linfoma de Hodgkin
Os “Sintomas B” representam um conjunto específico de manifestações sistêmicas com significativa relevância clínica e prognóstica no manejo do Linfoma de Hodgkin (LH). Sua correta identificação é imperativa para o estadiamento preciso da doença, a avaliação prognóstica e o delineamento da estratégia terapêutica.
Definição Criteriosa dos Sintomas B
A classificação de um paciente como portador de sintomas B requer a presença de um ou mais dos seguintes critérios, ocorrendo sem causa infecciosa ou outra etiologia identificável:
- Febre inexplicada: Temperatura corporal consistentemente superior a 38°C. Algumas definições especificam a necessidade de duração mínima (e.g., por mais de três dias consecutivos ou por mais de duas semanas).
- Sudorese noturna profusa: Transpiração noturna de intensidade suficiente para requerer a troca da roupa de cama ou do vestuário do paciente.
- Perda de peso inexplicada: Redução involuntária do peso corporal superior a 10% do peso basal nos últimos seis meses que antecederam o diagnóstico.
Significado Clínico e Implicações Prognósticas
A presença de sintomas B no LH é um marcador clínico de maior atividade metabólica tumoral e frequentemente se associa a uma carga tumoral mais elevada e a estágios mais avançados da doença ao diagnóstico. Estima-se que cerca de um terço dos pacientes com LH apresentem esses sintomas sistêmicos.
Os sintomas B são reconhecidos como um fator prognóstico adverso independente no Linfoma de Hodgkin. Sua presença correlaciona-se, em geral, com um prognóstico menos favorável e pode impactar negativamente as taxas de sobrevida. É importante ressaltar que os sintomas B são um dos diversos fatores prognósticos considerados na avaliação do LH, que incluem também o estágio de Ann Arbor, idade, níveis séricos de albumina, hemoglobina, contagem de leucócitos e linfócitos, e número de sítios extranodais acometidos.
Relevância no Estadiamento e na Conduta Terapêutica
A avaliação da presença ou ausência de sintomas B é um componente mandatório do estadiamento do LH, utilizando sistemas como Ann Arbor (modificado por Cotswolds ou Lugano). A presença destes sintomas categoriza o paciente como ‘B’ (e.g., Estágio IIB), enquanto a ausência o classifica como ‘A’ (e.g., Estágio IIA). A designação ‘B’ reflete uma maior agressividade biológica intrínseca da doença.
Consequentemente, a constatação de sintomas B influencia diretamente as decisões terapêuticas. Pacientes classificados como ‘B’ frequentemente requerem abordagens terapêuticas mais intensivas, como a utilização de regimes quimioterápicos mais potentes ou a consideração de modalidades terapêuticas adicionais, com o objetivo de mitigar o impacto prognóstico negativo associado a estas manifestações. A investigação e documentação meticulosa dos sintomas B são, portanto, cruciais na avaliação inicial e no seguimento de pacientes com Linfoma de Hodgkin.
Patologia I: A Célula de Reed-Sternberg e o Microambiente Tumoral
A identificação histopatológica da célula de Reed-Sternberg (CRS) é um pilar no diagnóstico do Linfoma de Hodgkin Clássico (LHc). Estas células neoplásicas, originadas de linfócitos B do centro germinativo ou pós-germinativos que escaparam da apoptose, são morfologicamente distintas, embora representem uma pequena fração da massa tumoral total.
Características Morfológicas da Célula de Reed-Sternberg Clássica
As CRS clássicas são células de grandes dimensões (15-45 μm), caracterizadas por um núcleo volumoso, frequentemente bilobulado ou multinucleado. Um traço marcante são os nucléolos proeminentes, eosinofílicos, muitas vezes circundados por um halo claro, o que lhes confere a aparência classicamente descrita como “olhos de coruja”. O citoplasma é geralmente abundante e pode variar de basofílico a anfofílico. Embora patognomônicas no contexto adequado, as CRS podem ser raras e de difícil localização no espécime histológico.
Variantes Morfológicas
- Células Lacunares: Uma variante observada predominantemente no subtipo Esclerose Nodular. Caracterizam-se por um artefato de retração do citoplasma durante a fixação em formalina, fazendo com que a célula pareça residir dentro de uma lacuna.
- Células Linfocíticas e Histiocíticas (L&H) ou ‘Células Pipoca’: Encontradas no Linfoma de Hodgkin com Predominância Linfocítica Nodular (LHPLN), uma entidade distinta do LHc. Estas células possuem núcleos multilobulados complexos e nucléolos menos proeminentes que as CRS clássicas.
Imunofenótipo e Origem Celular
A imuno-histoquímica é indispensável para confirmar a natureza das CRS no LHc. O perfil imunofenotípico clássico inclui forte positividade para CD30 e, na maioria dos casos, para CD15. Tipicamente, as CRS são negativas para o marcador pan-leucocitário CD45 (LCA) e para marcadores de linhagem B como CD20 e CD79a, apesar de sua origem em linfócitos B. Algumas variantes podem, no entanto, expressar certos marcadores B. A célula de Reed-Sternberg demonstra ineficiência na apresentação de antígenos, um fator que contribui para sua interação com o microambiente.
O Microambiente Tumoral Reativo
Uma característica distintiva do LHc é o extenso microambiente reativo que constitui a maior parte da massa tumoral (>98%). As CRS, embora sendo as células neoplásicas definidoras, estão imersas neste infiltrado celular inflamatório diversificado, que elas próprias recrutam e modulam ativamente através da secreção de um perfil complexo de citocinas e quimiocinas.
Este microambiente é composto por uma mistura heterogênea de células não neoplásicas, incluindo linfócitos T (frequentemente predominantes), linfócitos B, eosinófilos, neutrófilos, plasmócitos, histiócitos e fibroblastos. A interação dinâmica e complexa entre as CRS e as células do microambiente é fundamental para a patogênese do LHc, influenciando a sobrevivência das células neoplásicas, o crescimento tumoral, a evasão imune e contribuindo significativamente para as manifestações clínicas e sistêmicas da doença.
Patologia II: Classificação Histológica do Linfoma de Hodgkin
O Linfoma de Hodgkin (LH) é classificado em duas entidades clínico-patológicas distintas, com base em características morfológicas, imunofenotípicas e clínicas: o Linfoma de Hodgkin Clássico (LHc) e o Linfoma de Hodgkin com Predominância Linfocitária Nodular (LHPLN).
Linfoma de Hodgkin com Predominância Linfocitária Nodular (LHPLN)
Representando uma minoria dos casos de LH, o LHPLN distingue-se pela presença das células variantes L&H (linfocíticas e histiocíticas), também conhecidas como “células pipoca”, inseridas em um fundo nodular predominantemente composto por linfócitos B reativos. Esta entidade possui características clínicas e imunofenotípicas próprias, distintas do LHc, e habitualmente apresenta um curso clínico mais indolente.
Linfoma de Hodgkin Clássico (LHc)
O LHc constitui a maioria dos casos de LH e caracteriza-se pela identificação das células de Reed-Sternberg (CRS) clássicas ou suas variantes dentro de um microambiente inflamatório reativo. É subdividido em quatro subtipos histológicos, cada um com implicações prognósticas e, por vezes, terapêuticas distintas:
- Esclerose Nodular (LHc-EN): É o subtipo mais frequente, comum em adolescentes e adultos jovens, com apresentação envolvendo frequentemente linfonodos cervicais e mediastinais. Histopatologicamente, é marcado pela presença de densas bandas de colágeno (fibrose) que delimitam nódulos de tecido linfoide tumoral. Dentro destes nódulos, encontram-se as células lacunares, uma variante morfológica das CRS, em meio ao infiltrado inflamatório.
- Celularidade Mista (LHc-CM): Caracteriza-se por um infiltrado celular difuso e polimórfico, contendo CRS clássicas dispersas entre uma mistura rica de células inflamatórias reativas (linfócitos, eosinófilos, neutrófilos, plasmócitos e histiócitos), sem a formação de bandas de esclerose proeminentes. Este subtipo está frequentemente associado à presença de sintomas B e à infecção pelo vírus Epstein-Barr (EBV).
- Rico em Linfócitos (LHc-RL): Um subtipo raro, definido por um fundo celular com predomínio acentuado de linfócitos reativos e um número escasso de CRS clássicas. Geralmente, associa-se a um prognóstico relativamente favorável.
- Depleção Linfocitária (LHc-DL): É o subtipo mais raro e considerado o mais agressivo do LHc. Histologicamente, demonstra uma notável escassez de linfócitos reativos e um número relativamente elevado de CRS, que podem apresentar pleomorfismo. Está frequentemente associado a pacientes idosos, indivíduos com imunodeficiência (como infecção pelo HIV), doença em estádio avançado e um prognóstico historicamente menos favorável.
A correta identificação e classificação histológica do subtipo de Linfoma de Hodgkin são etapas fundamentais no manejo do paciente, influenciando diretamente a avaliação prognóstica e a estratégia terapêutica a ser adotada.
Diagnóstico e Padrões de Disseminação
O diagnóstico definitivo do Linfoma de Hodgkin (LH) é estabelecido mediante análise histopatológica de tecido linfoide afetado. A abordagem diagnóstica padrão-ouro requer uma biópsia excisional, que consiste na remoção completa de um linfonodo suspeito. Esta técnica é preferível às biópsias incisionais ou à Punção Aspirativa por Agulha Fina (PAAF), pois a PAAF, embora possa levantar a suspeita, frequentemente não fornece material suficiente para a caracterização histológica completa e a avaliação da arquitetura nodal, essencial para identificar as células de Reed-Sternberg (CRS) no seu contexto.
Indicações Clínicas para Biópsia Linfonodal
A indicação de biópsia de linfonodo deve ser considerada em situações clínicas específicas, tais como:
- Linfadenopatias que persistem por mais de 2 a 4 semanas, sem etiologia infecciosa ou inflamatória clara.
- Linfonodos que exibem características suspeitas de malignidade ao exame físico, como crescimento progressivo, consistência endurecida ou pétrea, e aderência a planos profundos.
- Adenomegalia em localizações de maior risco para malignidade, como as regiões supraclavicular ou epitroclear.
- Presença de linfadenopatia associada a sintomas B (febre inexplicada, sudorese noturna profusa, perda ponderal significativa e inexplicada).
- Identificação de linfadenopatia em pacientes com histórico pessoal de neoplasia.
A análise imuno-histoquímica do material obtido pela biópsia é indispensável para confirmar o fenótipo das células neoplásicas e para realizar o diagnóstico diferencial com outras doenças linfoproliferativas e condições reativas que podem mimetizar o LH.
Padrões de Disseminação
O Linfoma de Hodgkin classicamente demonstra um padrão previsível de disseminação linfática, ocorrendo por contiguidade. A doença tende a progredir de forma ordenada, avançando de um grupo de linfonodos acometido para os grupos anatomicamente adjacentes. Este padrão de disseminação diferencia o LH da maioria dos Linfomas Não Hodgkin (LNH), que frequentemente exibem padrões de disseminação mais variáveis e imprevisíveis, com maior propensão ao acometimento extranodal precoce. O envolvimento de órgãos extranodais no LH, embora caracterize a doença em estágio avançado (Estádio IV de Ann Arbor), é considerado menos comum nas fases iniciais comparativamente aos LNH.
Estadiamento: Sistemas Ann Arbor e Lugano
A determinação precisa da extensão anatômica do Linfoma de Hodgkin (LH) é um passo fundamental que antecede a decisão terapêutica e a avaliação prognóstica. O estadiamento é, portanto, uma ferramenta essencial para o planejamento do tratamento e para a predição da evolução clínica. Os sistemas de estadiamento, como o de Ann Arbor e suas modificações posteriores, incluindo as recomendações de Lugano, fornecem a estrutura para essa classificação, sendo cruciais para determinar a abordagem terapêutica.
Sistema de Estadiamento de Ann Arbor Modificado
O sistema de Ann Arbor, com suas modificações (como as do Consenso de Cotswolds), é a classificação clássica para a extensão do LH. Baseia-se na distribuição da doença em relação ao diafragma e no envolvimento de sítios extranodais. Os estágios são definidos como:
- Estádio I: Envolvimento de uma única região de linfonodos ou de uma única estrutura linfoide (ex: baço, timo), ou envolvimento localizado de um único órgão ou sítio extralinfático (IE).
- Estádio II: Envolvimento de duas ou mais regiões de linfonodos no mesmo lado do diafragma (acima ou abaixo).
- Estádio III: Envolvimento de regiões de linfonodos em ambos os lados do diafragma. Pode incluir o envolvimento do baço (IIIS).
- Estádio IV: Doença disseminada com envolvimento difuso ou disseminado de um ou mais órgãos extralinfáticos (como medula óssea, fígado ou pulmão), com ou sem envolvimento linfonodal associado.
Modificadores adicionais refinam o estadiamento e o prognóstico:
- A ou B: Indica a ausência (A) ou presença (B) dos ‘Sintomas B’ (febre inexplicada >38°C, sudorese noturna profusa, perda de peso >10% em 6 meses), cujo significado clínico e prognóstico foi detalhado na seção anterior. A presença de sintomas B geralmente indica uma doença mais ativa e impacta nas decisões terapêuticas.
- E: Indica envolvimento de um sítio extranodal por extensão direta ou contíguo/proximal a um sítio nodal conhecido de doença.
- X: Indica a presença de doença volumosa (bulky disease), classicamente definida como uma massa tumoral com diâmetro máximo superior a 10 cm ou uma massa mediastinal com largura maior que um terço da largura do tórax no mesmo nível em exames de imagem.
Classificação de Lugano e Abordagem Multimodal
A classificação de Lugano representa uma evolução e refinamento do sistema de Ann Arbor, destacando-se por incorporar formalmente os achados da Tomografia por Emissão de Pósitrons com Tomografia Computadorizada (PET-CT). Esta ferramenta é fundamental não apenas para um estadiamento inicial mais acurado, mas também para a avaliação da resposta ao tratamento, permitindo uma melhor definição da atividade metabólica da doença.
O processo de estadiamento completo requer uma abordagem integrada:
- Avaliação Clínica: Anamnese detalhada para identificar sintomas B e outros como prurido, e exame físico completo para avaliar as características das linfadenopatias (tamanho, consistência, localização) e detectar hepatoesplenomegalia ou outros sinais de envolvimento sistêmico.
- Exames de Imagem: A Tomografia Computadorizada (TC) de pescoço, tórax, abdômen e pelve, e principalmente a PET-CT, são essenciais para mapear o envolvimento linfonodal em todas as cadeias e identificar o acometimento de órgãos extranodais.
- Avaliação da Medula Óssea: Embora a PET-CT tenha alta sensibilidade para detectar envolvimento medular, a biópsia de medula óssea ainda pode ser considerada em cenários específicos, conforme as diretrizes de Lugano, especialmente em estágios avançados ou na presença de sintomas B.
Em suma, o estadiamento rigoroso, utilizando sistemas como Ann Arbor modificado e as diretrizes de Lugano, integrando dados clínicos, de imagem avançada e, quando indicado, avaliação da medula óssea, é indispensável para a estratificação de risco, planejamento terapêutico individualizado e monitoramento da resposta no Linfoma de Hodgkin.
Fatores Prognósticos e Visão Geral do Tratamento
A avaliação prognóstica e a definição da estratégia terapêutica no Linfoma de Hodgkin (LH) baseiam-se na integração de múltiplos fatores identificados durante o diagnóstico e estadiamento. A estratificação de risco resultante é fundamental para ajustar a intensidade do tratamento e prever a evolução clínica.
Fatores Prognósticos
Diversos fatores influenciam o prognóstico no Linfoma de Hodgkin, para além do estágio da doença determinado pelos sistemas de Ann Arbor ou Lugano. Estes incluem:
- Extensão da Doença (Estadiamento): Estágios mais avançados (III/IV) conferem um prognóstico menos favorável que os estágios iniciais (I/II).
- Presença de Sintomas B: A ocorrência de febre inexplicada, sudorese noturna profusa ou perda ponderal significativa (>10% em 6 meses) é um marcador de maior atividade biológica e um fator prognóstico adverso estabelecido.
- Idade: Pacientes com idade mais avançada no momento do diagnóstico tendem a apresentar um prognóstico menos favorável.
- Envolvimento Extranodal: O número de sítios extranodais acometidos e a presença de doença disseminada (Estágio IV) impactam negativamente o prognóstico.
- Doença Volumosa (‘Bulky Disease’): A presença de massa tumoral com diâmetro superior a 10 cm ou envolvimento mediastinal extenso (‘X’) é um indicador de pior prognóstico.
- Parâmetros Laboratoriais: Níveis séricos de albumina diminuídos, níveis de hemoglobina reduzidos, leucocitose e linfopenia são reconhecidos como fatores prognósticos adversos.
- Subtipo Histológico: Embora o tratamento tenha melhorado os resultados globais, o subtipo Depleção Linfocitária pode associar-se a um prognóstico mais reservado, enquanto o subtipo Rico em Linfócitos e o LH com Predominância Linfocítica Nodular (LHPLN) tendem a ter um curso mais favorável ou indolente. A Esclerose Nodular, mais comum, geralmente apresenta bom prognóstico em estágios iniciais.
- Status EBV: A presença do vírus Epstein-Barr nas células de Reed-Sternberg pode ter implicações prognósticas em determinados contextos clínicos e subtipos.
A integração destes fatores, frequentemente através de escores prognósticos validados, permite uma estratificação de risco mais precisa, orientando as decisões terapêuticas individualizadas.
Visão Geral do Tratamento
O Linfoma de Hodgkin é considerado uma neoplasia com altas taxas de curabilidade, especialmente em estágios iniciais. A escolha da abordagem terapêutica é multifatorial, dependendo do estadiamento preciso, dos fatores de risco identificados, do subtipo histológico e das condições clínicas gerais do paciente, seguindo diretrizes clínicas e protocolos estabelecidos.
A quimioterapia combinada é a base do tratamento. Regimes como o ABVD (Adriamicina, Bleomicina, Vimblastina, Dacarbazina) são o padrão para muitos pacientes, especialmente em doença inicial favorável. Para doença avançada ou de alto risco, regimes mais intensos como o BEACOPP escalonado (Bleomicina, Etoposídeo, Adriamicina, Ciclofosfamida, Vincristina, Procarbazina, Prednisona) podem ser considerados para otimizar os resultados, embora com maior perfil de toxicidade.
A radioterapia desempenha um papel importante, sobretudo como terapia de consolidação após a quimioterapia, visando erradicar doença residual em áreas específicas. Sua indicação e planejamento são adaptados com base no estágio inicial, na resposta metabólica avaliada por PET-CT e nos protocolos terapêuticos.
No cenário de doença recidivada ou refratária ao tratamento inicial, o transplante autólogo de células-tronco hematopoiéticas é a abordagem padrão com intenção curativa. Terapias mais recentes, como o anticorpo conjugado a droga brentuximabe vedotina (anti-CD30) e os inibidores de checkpoint imunológico (ex: nivolumabe, pembrolizumabe), representam avanços significativos para esses casos.
É importante notar que o manejo do LHPLN pode diferir do LH Clássico, refletindo sua biologia e curso clínico distintos. Ademais, o acompanhamento de longo prazo é essencial para monitorar e gerenciar potenciais efeitos colaterais tardios associados aos tratamentos.