A Hiperplasia Adrenal Congênita (HAC) compreende um grupo heterogêneo de doenças genéticas de herança autossômica recessiva, caracterizadas por defeitos enzimáticos na via de biossíntese dos hormônios esteroides no córtex adrenal. Estes defeitos resultam fundamentalmente na produção inadequada de cortisol e, em diversos graus, de aldosterona. Este artigo explora a etiologia, genética e fisiopatologia da HAC por deficiência de 21-hidroxilase (CYP21A2), abordando vias hormonais, feedback e suas consequências.
Deficiência de 21-Hidroxilase (21OHD): A Causa Mais Comum da HAC
Dentre as diferentes formas de HAC, a deficiência da enzima 21-hidroxilase (21OHD) se destaca como a causa mais comum, respondendo por aproximadamente 90% a 95% de todos os casos diagnosticados. Esta condição é decorrente de mutações no gene CYP21A2, localizado no cromossomo 6p21.3, responsável pela codificação da enzima 21-hidroxilase. A herança autossômica recessiva implica que um indivíduo afetado herda duas cópias mutadas do gene CYP21A2, uma de cada progenitor, que usualmente são portadores heterozigotos assintomáticos.
Fisiopatologia: O Bloqueio Enzimático e Suas Consequências Bioquímicas
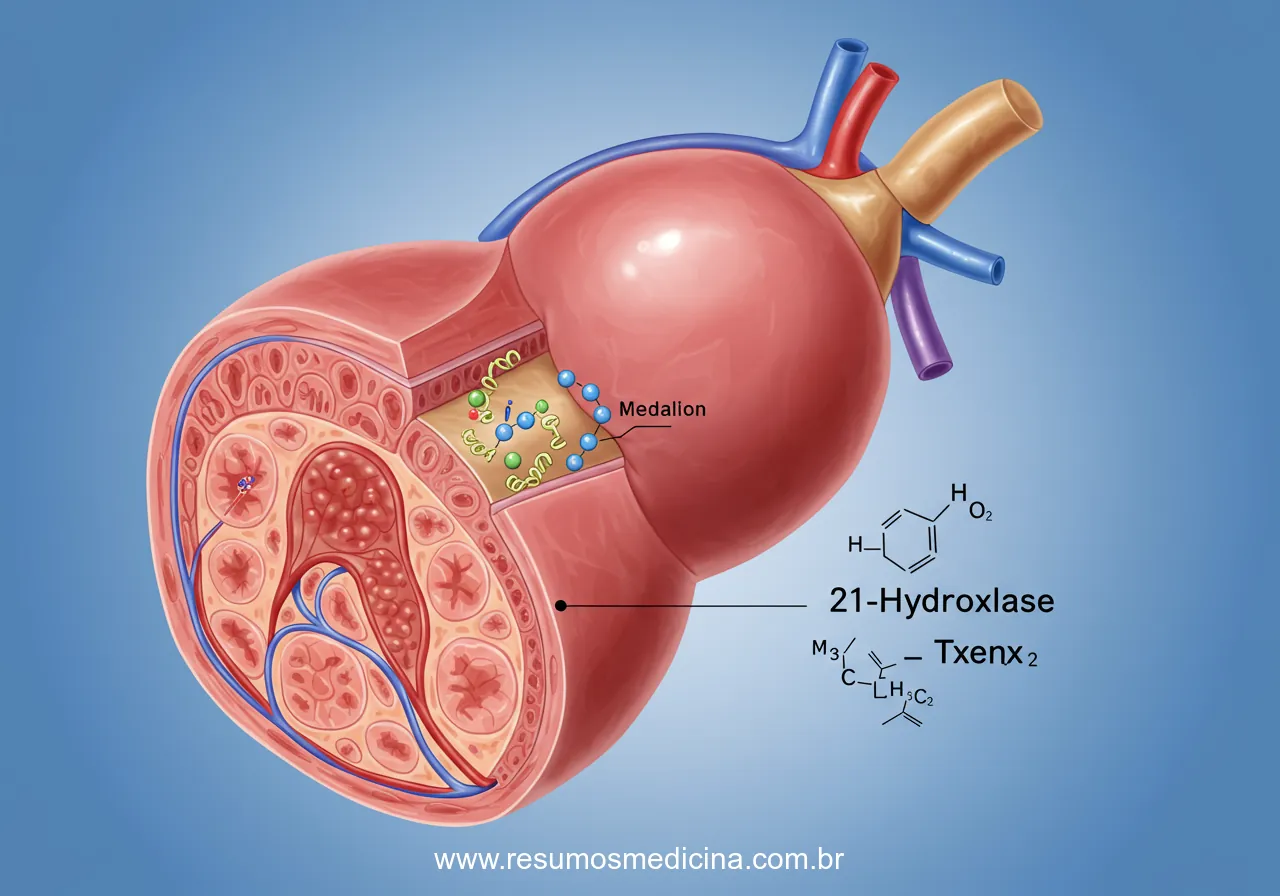
A fisiopatologia central da HAC por deficiência de 21-hidroxilase reside na interrupção de etapas enzimáticas cruciais na esteroidogênese adrenal. A enzima 21-hidroxilase (CYP21A2) desempenha um papel indispensável na biossíntese de glicocorticoides e mineralocorticoides. Especificamente, ela catalisa a hidroxilação na posição 21 de dois substratos fundamentais:
- A conversão da progesterona em desoxicorticosterona (DOC), um precursor essencial na via de síntese da aldosterona.
- A conversão da 17-hidroxiprogesterona (17-OHP) em 11-desoxicortisol, um intermediário vital na via de síntese do cortisol.
A deficiência ou ausência da atividade da 21-hidroxilase, decorrente de mutações no gene CYP21A2, impede a conversão eficiente desses substratos. Este bloqueio enzimático tem duas consequências bioquímicas primárias:
- Diminuição da produção de hormônios finais: Ocorre uma redução significativa na síntese de cortisol e, dependendo da gravidade da mutação, a produção de aldosterona também pode ser comprometida, levando à deficiência de mineralocorticoides.
- Acúmulo de precursores a montante: Os substratos que precedem o bloqueio enzimático acumulam-se nas glândulas adrenais e na circulação. O precursor mais notavelmente acumulado é a 17-hidroxiprogesterona (17-OHP). Seus níveis elevados são uma característica bioquímica distintiva da deficiência de 21-hidroxilase e servem como um marcador diagnóstico fundamental para a condição. Outros precursores, como a progesterona, também podem se acumular.
Esse acúmulo não apenas evidencia o local do defeito metabólico, mas também direciona o fluxo esteroidogênico para vias alternativas, notadamente a produção excessiva de andrógenos adrenais.
Desregulação do Eixo HHA e Hiperplasia Adrenal
Na HAC por deficiência de 21-hidroxilase, a dinâmica do eixo Hipotálamo-Hipófise-Adrenal (HHA) é significativamente alterada devido à produção insuficiente de cortisol, que desempenha um papel crucial na regulação do próprio eixo através de um mecanismo de feedback negativo. A deficiência da enzima 21-hidroxilase impede a síntese eficaz de cortisol, resultando na perda ou redução desse feedback negativo sobre o hipotálamo e a hipófise.
Com a atenuação do freio inibitório exercido pelo cortisol, ocorre um aumento compensatório na secreção de CRH e, subsequentemente, de ACTH. A hipófise libera quantidades elevadas de ACTH na tentativa de estimular as glândulas adrenais a produzir mais cortisol e superar o bloqueio enzimático. Entretanto, devido à deficiência enzimática persistente, o estímulo crônico e excessivo pelo ACTH não eleva a produção de cortisol, induzindo um aumento no tamanho das glândulas adrenais, processo denominado hiperplasia adrenal. Adicionalmente, o ACTH elevado impulsiona a esteroidogênese adrenal a montante do bloqueio, resultando no acúmulo de precursores como a 17-hidroxiprogesterona e no desvio dessas moléculas para a via de síntese de andrógenos.
Portanto, a hiperplasia adrenal observada na HAC por deficiência de 21-hidroxilase é uma consequência direta da estimulação adrenocortical crônica por níveis elevados de ACTH, decorrentes da falha no mecanismo de feedback negativo devido à produção insuficiente de cortisol.
Desvio Metabólico para a Produção de Andrógenos e Virilização
Na HAC por deficiência de 21-hidroxilase, a atividade reduzida ou ausente desta enzima crucial impede a conversão fisiológica de precursores esteroides nas vias de síntese de cortisol e aldosterona. Este bloqueio enzimático resulta inevitavelmente no acúmulo de substratos a montante, com destaque para a 17-OHP, que são compulsoriamente desviados para a via metabólica alternativa da produção de andrógenos adrenais.
Consequentemente, observa-se um aumento na produção de andrógenos como testosterona, di-hidroepiandrosterona (DHEA) e androstenediona. Esse aumento é ainda exacerbado pela elevação secundária dos níveis de hormônio adrenocorticotrófico (ACTH), que estimula cronicamente o córtex adrenal hiperplásico.
Manifestações Clínicas do Hiperandrogenismo
O excesso de andrógenos resultante deste desvio metabólico é o principal responsável pelas manifestações clínicas de virilização e outras alterações associadas ao hiperandrogenismo na HAC por deficiência de 21-hidroxilase:
- Em indivíduos com cariótipo 46,XX: A exposição intrauterina a níveis elevados de andrógenos durante o desenvolvimento fetal leva à virilização da genitália externa, manifestando-se como genitália ambígua, incluindo hipertrofia do clitóris (clitoromegalia) e diferentes graus de fusão dos grandes lábios (fusão labial). É fundamental ressaltar que os órgãos genitais internos femininos (útero, trompas e ovários) permanecem intactos.
- Em ambos os sexos: O hiperandrogenismo pós-natal pode causar o desenvolvimento precoce de características sexuais secundárias, como pubarca precoce (aparecimento de pelos pubianos e axilares), acne e odor axilar apócrino. Observa-se também uma aceleração da velocidade de crescimento e um avanço da idade óssea. Se não houver intervenção terapêutica adequada, esse avanço pode levar ao fechamento prematuro das epífises ósseas, resultando em baixa estatura na vida adulta. A puberdade precoce central também pode ser desencadeada.
- Em indivíduos com cariótipo 46,XY: Embora a genitália externa seja tipicamente masculina ao nascimento, pode haver sinais como aumento do tamanho do pênis e hiperpigmentação escrotal. São igualmente suscetíveis à pubarca precoce, aceleração do crescimento e avanço da idade óssea.
Portanto, o redirecionamento da esteroidogênese para a produção excessiva de andrógenos é um evento fisiopatológico central na deficiência de 21-hidroxilase, determinando as manifestações de virilização e os distúrbios do desenvolvimento puberal característicos desta forma de HAC.
Consequências da Deficiência de Mineralocorticoides: Perda de Sal e Crise Adrenal
Nas formas clássicas da HAC por deficiência da enzima 21-hidroxilase (CYP21A2), além da produção inadequada de cortisol, frequentemente ocorre um comprometimento significativo na síntese de aldosterona, que desempenha um papel crucial na homeostase hidroeletrolítica. A deficiência de 21-hidroxilase impede a conversão eficiente de progesterona em desoxicorticosterona, um precursor essencial na via de produção da aldosterona.
Manifestações da Deficiência de Aldosterona
As consequências fisiopatológicas diretas da deficiência de aldosterona incluem:
- Comprometimento da Reabsorção de Sódio: A falta de ação da aldosterona nos túbulos renais leva a uma diminuição na reabsorção de sódio, resultando em perda urinária excessiva deste íon (perda de sal). Clinicamente, isso se manifesta como hiponatremia.
- Retenção de Potássio: Simultaneamente, a excreção de potássio é reduzida, levando ao seu acúmulo no organismo e, consequentemente, à hipercalemia. Níveis elevados de potássio sérico representam um risco substancial, incluindo a possibilidade de arritmias cardíacas potencialmente fatais.
- Desidratação e Hipovolemia: A perda renal de sódio é acompanhada por uma perda osmótica de água, conduzindo à desidratação e contração do volume intravascular (hipovolemia).
- Hipotensão e Choque Hipovolêmico: A hipovolemia resultante pode causar hipotensão arterial. Em casos graves e não tratados, essa condição pode progredir rapidamente para choque hipovolêmico.
Crise Adrenal (Crise Perdedora de Sal)
A combinação severa de hiponatremia, hipercalemia, desidratação, hipovolemia e hipotensão caracteriza a crise adrenal ou crise perdedora de sal. Esta é uma emergência médica aguda, especialmente crítica e de alto risco no período neonatal, podendo ser fatal se não diagnosticada e tratada prontamente. A deficiência concomitante de cortisol, também presente na HAC clássica, contribui adicionalmente para a instabilidade hemodinâmica, o risco de hipoglicemia e a gravidade geral da crise adrenal.
Impacto da Deficiência de Glicocorticoides: Hipoglicemia e Resposta ao Estresse
A deficiência de cortisol na HAC por deficiência de 21-hidroxilase acarreta consequências fisiopatológicas significativas, comprometendo a homeostase metabólica e a capacidade de resposta a desafios fisiológicos.
Comprometimento da Resposta ao Estresse e Risco de Crise Adrenal
O cortisol é um hormônio fundamental na resposta fisiológica ao estresse. Sua ausência ou produção inadequada, como ocorre na HAC, resulta em uma resposta inadequada a situações de estresse físico ou metabólico. Essa vulnerabilidade torna os pacientes suscetíveis à crise adrenal, uma emergência médica potencialmente fatal caracterizada por hipotensão, hipoglicemia e choque. A insuficiência adrenal global, frequentemente envolvendo tanto a deficiência de cortisol quanto a de aldosterona, exacerba a instabilidade hemodinâmica e eleva substancialmente o risco de crise adrenal.
Desregulação da Glicemia e Hipoglicemia
O cortisol exerce um papel crucial na manutenção da normoglicemia. Na HAC, a deficiência de cortisol compromete diretamente os processos metabólicos vitais, como a estimulação da gliconeogênese hepática e da glicogenólise. A incapacidade de aumentar adequadamente a produção de glicose endógena, particularmente durante períodos de jejum ou durante situações de estresse (como infecções ou cirurgias), predispõe os indivíduos afetados a episódios de hipoglicemia.
Conclusão
A Hiperplasia Adrenal Congênita por deficiência de 21-hidroxilase é uma condição complexa, resultante de um bloqueio enzimático na esteroidogênese adrenal. A deficiência de cortisol e, em muitos casos, de aldosterona, juntamente com o excesso de andrógenos, leva a uma variedade de manifestações clínicas que podem variar em severidade. O diagnóstico precoce e o manejo adequado são cruciais para minimizar as complicações a longo prazo e garantir a qualidade de vida dos pacientes afetados.