O estudo da interação entre fatores genéticos e o desenvolvimento de neoplasias é fundamental para a oncologia moderna. A compreensão dos mecanismos de hereditariedade no câncer permite não apenas a identificação de indivíduos e famílias com risco aumentado, mas também a implementação de estratégias personalizadas de vigilância e prevenção, com impacto direto na morbimortalidade associada a diversas síndromes oncológicas.
Introdução à Predisposição Hereditária ao Câncer
A predisposição hereditária ao câncer refere-se à herança de mutações germinativas em genes específicos que conferem um risco significativamente elevado para o desenvolvimento de neoplasias. Embora a maioria dos casos de câncer seja esporádica, uma proporção clinicamente relevante, estimada em cerca de 10% para câncer de mama, por exemplo, decorre dessa suscetibilidade genética herdada. Essas predisposições frequentemente envolvem mutações em genes supressores de tumor, como os emblemáticos BRCA1 e BRCA2. Tais genes desempenham papéis cruciais na manutenção da estabilidade genômica, participando ativamente em vias de reparo do DNA, como a recombinação homóloga para correção de quebras de fita dupla, e na indução de apoptose (morte celular programada) em células com danos genômicos irreparáveis. A perda de função desses genes, decorrente das mutações herdadas, compromete esses mecanismos de salvaguarda celular, levando ao acúmulo de alterações genéticas e aumentando a probabilidade de transformação maligna.
O padrão de herança mais comum para essas síndromes é o autossômico dominante. Isso significa que a herança de apenas uma cópia mutada do gene (proveniente de um dos genitores) é suficiente para aumentar o risco de câncer, e cada descendente de um portador tem 50% de chance de herdar a mutação. Contudo, a penetrância dessas mutações – a proporção de portadores que efetivamente desenvolverão a doença – é frequentemente alta, mas incompleta. Isso implica que, embora o risco seja substancialmente elevado, nem todos os indivíduos com a mutação desenvolverão câncer, e a manifestação pode ser influenciada por outros fatores genéticos (genes modificadores) e ambientais. A identificação precisa de indivíduos em risco, através da avaliação criteriosa do histórico pessoal e familiar (detalhando tipos de câncer, idade de diagnóstico em parentes de primeiro, segundo e terceiro graus) e do aconselhamento genético, é crucial. Este processo permite a discussão informada sobre testes genéticos e a implementação de estratégias individualizadas de manejo, que podem incluir vigilância intensificada, quimioprevenção ou cirurgias redutoras de risco.
Principais Abordagens e Conceitos Fundamentais
- Avaliação de Risco e Histórico Familiar: Coleta detalhada de informações sobre múltiplos parentes (primeiro, segundo, terceiro graus), tipos de câncer, idade de diagnóstico e ancestralidade (como a judaica Ashkenazi, com maior prevalência de mutações específicas como BRCA1/2) para identificar padrões sugestivos de hereditariedade.
- Aconselhamento Genético: Processo essencial que envolve a educação do paciente sobre riscos genéticos, discussão dos benefícios e limitações dos testes genéticos, interpretação de resultados e orientação sobre as opções de manejo clínico personalizadas e suas implicações.
- Estratégias de Manejo Personalizado: Com base no risco identificado e/ou resultado genético, incluem: (1) Vigilância Intensificada (rastreamento mais precoce, frequente ou com modalidades adicionais, como RM de mamas), (2) Quimioprevenção (uso de fármacos como tamoxifeno ou inibidores da aromatase para reduzir risco de certos cânceres) e (3) Cirurgia Redutora de Risco (como mastectomia profilática e salpingo-ooforectomia bilateral).
Bases Genéticas da Hereditariedade no Câncer: Genes Supressores, Herança e Penetrância
A compreensão dos mecanismos genéticos subjacentes à predisposição hereditária ao câncer é fundamental para a avaliação de risco e o manejo clínico adequado. Fatores como a função dos genes envolvidos, o padrão de herança e a penetrância das mutações determinam o fenótipo clínico.
O Papel Crucial dos Genes Supressores de Tumor
Genes supressores de tumor são essenciais para a manutenção da integridade genômica e o controle celular. Eles regulam o ciclo celular, promovem o reparo de danos no DNA e, crucialmente, induzem a apoptose (morte celular programada) em células com danos irreparáveis ou potencial tumorigênico. A perda de função desses genes, decorrente de mutações germinativas, é um evento chave na carcinogênese, permitindo que células anormais proliferem descontroladamente.
- Genes BRCA1 e BRCA2: Codificam proteínas envolvidas no reparo de quebras de fita dupla do DNA através da via de recombinação homóloga, além de participarem da indução de apoptose. Mutações nesses genes comprometem a capacidade de reparo, levando ao acúmulo de danos genômicos e aumentando significativamente o risco de câncer de mama, ovário, próstata e pâncreas.
- Gene TP53: Codifica a proteína p53, um fator de transcrição central na resposta ao estresse celular. A p53 regula a progressão do ciclo celular, o reparo do DNA, a senescência e a apoptose. Mutações germinativas no TP53 causam a Síndrome de Li-Fraumeni e são as alterações genéticas mais comuns encontradas em diversos cânceres humanos, resultando na perda da função supressora e contribuindo para a instabilidade genômica e tumorigênese.
Padrão de Herança Autossômica Dominante
Muitas síndromes de predisposição hereditária ao câncer, incluindo as associadas a mutações nos genes BRCA1, BRCA2, TP53 (Síndrome de Li-Fraumeni), PTEN (Síndrome de Cowden) e CDH1 (Câncer Gástrico Difuso Hereditário – CGDH), seguem um padrão de herança autossômica dominante. Neste modelo, a herança de apenas uma cópia mutada do gene (de um dos pais) é suficiente para conferir um risco aumentado de desenvolver a doença. Cada descendente de um portador tem 50% de probabilidade de herdar a mutação e o risco associado.
Penetrância e Expressividade Variável
Penetrância define a proporção de indivíduos portadores de uma mutação específica que manifestam o fenótipo associado (neste caso, o câncer). Nas síndromes hereditárias, a penetrância é frequentemente alta, mas incompleta. Por exemplo, nem todos os portadores de mutações em BRCA1 ou BRCA2 desenvolverão câncer.
- Estimativas de Risco para BRCA1/2: O risco cumulativo de câncer de mama até os 80 anos é estimado em cerca de 72% para portadoras de mutação em BRCA1 e 69% para BRCA2. O risco de câncer de ovário até os 80 anos é de aproximadamente 44% para BRCA1 e 17% para BRCA2. Notavelmente, cânceres de mama associados a mutações BRCA1 tendem a se manifestar em idades mais jovens, frequentemente entre 30 e 40 anos.
- Fatores de Modificação: A penetrância incompleta e a expressividade variável (variação na idade de início, tipo ou gravidade do câncer entre portadores da mesma mutação) são influenciadas por múltiplos fatores. Estes incluem o gene específico envolvido (BRCA1 vs. BRCA2), a localização exata da mutação dentro do gene, a presença de outros genes modificadores, o histórico familiar, a etnia, fatores ambientais e o estilo de vida. Essa complexidade reforça a necessidade de avaliação de risco e planejamento de manejo individualizados.
Síndrome de Câncer de Mama e Ovário Hereditário (HBOC): Foco nos Genes BRCA1 e BRCA2
A Síndrome de Câncer de Mama e Ovário Hereditário (HBOC) é a forma mais comum e bem caracterizada de predisposição hereditária a essas neoplasias, sendo majoritariamente causada por mutações germinativas nos genes supressores de tumor BRCA1 e BRCA2. Estima-se que aproximadamente metade dos casos de câncer de mama e ovário de natureza hereditária esteja associada a mutações nesses dois genes.
Conforme abordado previamente, a função essencial dos genes BRCA1 e BRCA2 reside na manutenção da estabilidade genômica, codificando proteínas cruciais para o reparo de quebras de fita dupla do DNA através da via de recombinação homóloga e para a indução de apoptose em células danificadas. Mutações patogênicas nesses genes comprometem essas funções, levando à instabilidade genômica e a um risco substancialmente elevado de desenvolvimento neoplásico, seguindo um padrão de herança autossômica dominante com alta penetrância, ainda que incompleta.
Prevalência e Aspectos Populacionais das Mutações BRCA1/2
Embora as mutações germinativas nos genes BRCA1 e BRCA2 sejam relativamente raras na população geral, sua prevalência é significativamente maior em subgrupos específicos. Indivíduos com histórico familiar robusto de câncer de mama ou ovário, especialmente com diagnóstico em idade precoce (antes dos 45-50 anos), e aqueles pertencentes a certas etnias, como os judeus Ashkenazi (onde mutações fundadoras específicas são mais comuns), apresentam uma probabilidade aumentada de serem portadores dessas mutações. A identificação da ancestralidade Ashkenazi é, portanto, um fator relevante na avaliação de risco e indicação de teste genético.
Espectro Oncológico e Características Clínico-Patológicas na HBOC
Indivíduos com mutações em BRCA1 ou BRCA2 enfrentam um risco aumentado para um espectro específico de neoplasias. Além do risco proeminente para câncer de mama e câncer epitelial de ovário (incluindo trompas de Falópio), há também um risco elevado para outras malignidades:
- Câncer de Próstata: Risco aumentado, particularmente para formas mais agressivas (escore de Gleason ≥ 7) e com diagnóstico em idade mais jovem.
- Câncer de Pâncreas: Risco elevado comparado à população geral.
- Câncer de Mama Masculino: Incidência significativamente maior em portadores de mutações BRCA, especialmente BRCA2.
Tumores associados a mutações BRCA1 frequentemente exibem características clínico-patológicas distintas. Tipicamente, são diagnosticados em idade mais jovem, apresentam alto grau histológico (indicando maior agressividade) e são mais propensos a serem negativos para receptores hormonais (estrogênio – RE e progesterona – RP). Como resultado, é comum que sejam classificados como tumores triplo-negativos (RE-negativo, RP-negativo e HER2-negativo), um subtipo molecular com implicações prognósticas e terapêuticas específicas. A associação de mutações BRCA1 com o risco de câncer de ovário também é notavelmente mais elevada do que a observada em mutações BRCA2.
Outras Síndromes Genéticas Associadas ao Risco de Câncer de Mama e Outros Tumores
Além da Síndrome de Câncer de Mama e Ovário Hereditário (HBOC) associada aos genes BRCA1 e BRCA2, diversas outras síndromes genéticas conferem predisposição a neoplasias, incluindo o câncer de mama. O reconhecimento destas síndromes é crucial para um aconselhamento genético preciso, definição de estratégias de rastreamento personalizadas e implementação de medidas de redução de risco apropriadas.
Síndrome de Li-Fraumeni (Mutação no Gene TP53)
A Síndrome de Li-Fraumeni é causada por mutações germinativas no gene TP53, que codifica a proteína p53, um fator de transcrição central na resposta ao estresse celular e frequentemente mutado em diversos cânceres humanos. A perda de função da p53 compromete mecanismos cruciais de supressão tumoral. Esta síndrome está associada a um espectro alargado de tumores, incluindo sarcomas, tumores cerebrais, carcinomas adrenocorticais e leucemias, além de um risco aumentado de câncer de mama, particularmente em idades precoces. Portadores de mutações TP53 podem necessitar de protocolos de vigilância intensiva e discussão sobre estratégias redutoras de risco específicas.
Síndrome de Cowden (Mutação no Gene PTEN)
Mutações germinativas no gene supressor de tumor PTEN causam a Síndrome de Cowden. Esta condição é caracterizada pelo desenvolvimento de múltiplos hamartomas em diversos órgãos e por um risco aumentado de neoplasias malignas, notadamente câncer de mama, tireoide (especialmente folicular) e endométrio. O reconhecimento das manifestações clínicas da Síndrome de Cowden é essencial para direcionar o teste genético para o gene PTEN e implementar o seguimento oncológico adequado para os tumores associados.
Síndrome de Lynch (Mutação em Genes de Reparo de Mismatch – MMR)
Embora primariamente associada ao câncer colorretal hereditário não poliposo (HNPCC) e ao câncer de endométrio, a Síndrome de Lynch, causada por mutações em genes do sistema de reparo de erros de replicação do DNA (MMR), também eleva o risco para outros tumores, incluindo câncer de ovário, estômago, trato urinário (pelve renal, ureter), entre outros. A deficiência no sistema MMR leva à Instabilidade de Microssatélites (MSI), um marcador molecular característico. Microssatélites são sequências repetitivas de DNA; sua instabilidade (MSI) refere-se a alterações no comprimento dessas sequências devido à falha no reparo de erros durante a replicação do DNA, podendo levar ao acúmulo de mutações em genes críticos. A suspeita clínica para investigação da Síndrome de Lynch pode ser guiada pelos Critérios de Amsterdam II, que focam no histórico familiar de cânceres associados, e pelos Critérios de Bethesda Revisados, que definem indicações para testar tumores (principalmente colorretais) para MSI com base em características clínico-patológicas.
Câncer Gástrico Hereditário Difuso (CGHD – Mutação no Gene CDH1)
Mutações germinativas no gene CDH1, que codifica a E-caderina, causam o Câncer Gástrico Hereditário Difuso (CGHD). A E-caderina é uma glicoproteína transmembranar essencial para a adesão célula-célula nas junções aderentes, crucial para a manutenção da polaridade celular, organização tecidual e supressão da invasão. A perda de sua função predispõe a um risco extremamente elevado de câncer gástrico do tipo difuso (conforme classificação de Lauren, caracterizado histologicamente por células pouco coesas, frequentemente com morfologia em anel de sinete, que se infiltram difusamente na parede gástrica) e também a um risco significativamente aumentado de carcinoma lobular invasivo da mama em mulheres. Critérios clínicos para suspeita de CGHD incluem múltiplos casos de câncer gástrico difuso na família, diagnóstico antes dos 40 anos, ou a ocorrência simultânea de câncer gástrico difuso e carcinoma lobular da mama na mesma família. Em mulheres portadoras de mutação no CDH1, recomenda-se rastreamento mamário intensificado, geralmente com exame clínico, mamografia anual e ressonância magnética mamária a partir dos 30 anos. Dada a alta penetrância e a natureza agressiva do câncer gástrico difuso, a gastrectomia total profilática é frequentemente recomendada para portadores de mutações patogênicas no CDH1 após avaliação multidisciplinar.
Avaliação de Risco e Critérios para Indicação de Testagem Genética
A identificação de indivíduos e famílias com risco aumentado para síndromes de câncer hereditário é um componente essencial da prática oncológica. O processo de avaliação de risco visa identificar aqueles com maior probabilidade de serem portadores de mutações germinativas, permitindo o aconselhamento genético adequado e a tomada de decisões informadas sobre testagem e manejo subsequente.
A Importância Fundamental do Histórico Familiar Detalhado
A coleta de um histórico familiar detalhado e abrangente é a pedra angular na avaliação do risco de câncer hereditário. Esta avaliação deve incluir informações sobre múltiplos graus de parentesco (primeiro, segundo e terceiro graus), detalhando os tipos de câncer diagnosticados em cada familiar, a idade ao diagnóstico e, se disponíveis, resultados de testes genéticos prévios. A presença de múltiplos casos de câncer, diagnósticos em idades precoces ou a ocorrência de tipos específicos de tumores na família aumentam a suspeita de uma predisposição hereditária. A magnitude do risco é influenciada pelo número de parentes afetados, o grau de parentesco e a idade ao diagnóstico. É crucial notar que, embora um histórico familiar positivo aumente significativamente a suspeita, a ausência de histórico familiar não exclui a possibilidade de uma mutação hereditária germinativa.
Critérios Clínicos para Indicação de Testagem Genética
A decisão de prosseguir com a testagem genética é orientada por critérios clínicos e familiares específicos que aumentam a probabilidade pré-teste de identificar uma mutação patogênica. Diretrizes clínicas consolidadas, como as da NCCN (National Comprehensive Cancer Network), fornecem critérios detalhados para essa indicação.
Síndrome de Câncer de Mama e Ovário Hereditário (HBOC) – Critérios BRCA1/2
A testagem para mutações nos genes BRCA1 e BRCA2 é recomendada em indivíduos que apresentam características pessoais ou familiares sugestivas, incluindo:
- Diagnóstico de câncer de mama em idade jovem (geralmente ≤45-50 anos).
- Diagnóstico de câncer de ovário epitelial (em qualquer idade), trompas de falópio ou peritoneal primário.
- Diagnóstico de câncer de mama triplo-negativo.
- Diagnóstico de câncer de mama bilateral.
- Ocorrência de câncer de mama e ovário no mesmo indivíduo.
- Diagnóstico de câncer de mama masculino.
- Histórico familiar com múltiplos casos (em parentes de 1º, 2º ou 3º graus) de câncer de mama, ovário, pâncreas ou próstata (especialmente agressivo, Gleason ≥ 7).
- Ancestralidade Judaica Ashkenazi, associada a uma maior prevalência de mutações fundadoras, especialmente na presença de histórico familiar de cânceres associados.
- Identificação prévia de uma mutação BRCA1/2 patogênica em um membro da família.
Síndrome de Lynch – Critérios de Bethesda Revisados para Teste de MSI
Para a suspeita de Síndrome de Lynch (Câncer Colorretal Hereditário Não Poliposo – HNPCC), os Critérios de Bethesda Revisados indicam quando um tumor colorretal (CCR) deve ser testado para Instabilidade de Microssatélites (MSI), um marcador molecular associado a esta síndrome:
- CCR diagnosticado em paciente com idade inferior a 50 anos.
- Presença de tumores colorretais sincrônicos ou metacrônicos, ou outros tumores associados à Síndrome de Lynch (ex: endometrial, estômago, ovário, pâncreas, ureter, pelve renal, cérebro, biliar, intestino delgado, sebáceo), independentemente da idade.
- CCR com histologia sugestiva de alta instabilidade de microssatélites (MSI-H) – como presença de linfócitos infiltrantes de tumor (TILs), reação estromal tipo Crohn-like, diferenciação mucinosa/anel de sinete ou medular – diagnosticado em paciente com idade inferior a 60 anos.
- CCR diagnosticado em paciente com um ou mais parentes de primeiro grau com câncer associado à Síndrome de Lynch, sendo um dos cânceres diagnosticado antes dos 50 anos.
- CCR diagnosticado em paciente com dois ou mais parentes de primeiro ou segundo grau com câncer associado à Síndrome de Lynch, independentemente da idade.
Os Critérios de Amsterdam II, embora focados primariamente na avaliação da história familiar para suspeita clínica de Síndrome de Lynch, complementam a avaliação e requerem a exclusão de Polipose Adenomatosa Familiar (PAF).
Câncer Gástrico Hereditário Difuso (CGHD) – Critérios de Suspeita
A suspeita clínica de CGHD, relacionada a mutações no gene CDH1, deve ser considerada nos seguintes cenários:
- Múltiplos casos de câncer gástrico do tipo difuso na família.
- Diagnóstico de câncer gástrico difuso em idade jovem (inferior a 40 anos).
- Ocorrência combinada de câncer gástrico difuso e carcinoma lobular invasivo da mama na mesma família ou indivíduo.
A aplicação rigorosa destes critérios é fundamental para selecionar os pacientes que mais se beneficiarão do encaminhamento para aconselhamento genético especializado. Este processo é crucial para discutir as implicações, benefícios e limitações da testagem genética e para implementar estratégias personalizadas de rastreamento e redução de risco, quando apropriado.
Aconselhamento Genético Oncológico: Processo, Objetivos e Importância Clínica
O aconselhamento genético oncológico é um processo de comunicação fundamental para indivíduos e famílias com suspeita ou confirmação de síndromes de predisposição hereditária ao câncer. Conduzido por profissionais especializados, visa fornecer informação, educação e apoio psicossocial, capacitando os envolvidos a tomar decisões autônomas sobre sua saúde.
Componentes Essenciais do Processo
O processo de aconselhamento genético é individualizado e tipicamente envolve sessões pré e pós-teste genético, abordando os seguintes objetivos:
- Avaliação e Contextualização do Risco: Integração e interpretação dos dados do histórico pessoal e familiar (obtidos durante a avaliação de risco inicial) para estimar a probabilidade individual de ser portador de uma mutação e o risco associado a síndromes específicas.
- Educação sobre Genética do Câncer: Explicação dos aspectos genéticos relevantes para o caso, incluindo os genes de suscetibilidade implicados, os padrões de herança e a penetrância das mutações, contextualizando o risco individualizado.
- Discussão Aprofundada sobre Testagem Genética: Análise das opções de testes disponíveis (desde análise de genes específicos, como BRCA1/BRCA2, até painéis multigênicos), ponderando os potenciais benefícios (informação para manejo clínico, orientação para familiares), limitações (possibilidade de resultados inconclusivos como Variantes de Significado Incerto – VUS, custo) e as indicações clínicas para a testagem com base nos critérios estabelecidos.
- Interpretação de Resultados: Comunicação clara e detalhada do significado dos resultados dos testes genéticos (sejam positivos para mutação patogênica, negativos ou VUS) e suas consequências diretas para as estratégias de manejo clínico do paciente e a avaliação de risco para familiares.
- Orientação sobre Manejo Clínico Personalizado: Discussão das opções de seguimento baseadas no risco genético identificado, incluindo estratégias de vigilância intensificada (com modalidades e intervalos específicos) e intervenções de redução de risco (como quimioprevenção e cirurgias profiláticas), considerando as diretrizes clínicas atuais e as preferências do paciente.
- Apoio Psicossocial: Abordagem das potenciais implicações emocionais, psicológicas e sociais associadas ao conhecimento do risco genético, ao processo de testagem e aos seus resultados, oferecendo suporte e direcionamento adequados.
Desta forma, o aconselhamento genético oncológico desempenha um papel crucial ao traduzir informações genéticas complexas em conhecimento clinicamente acionável, permitindo que pacientes e famílias compreendam seu risco e participem ativamente das decisões sobre as melhores estratégias de prevenção, rastreamento e tratamento, alinhadas aos seus valores e preferências.
Estratégias de Rastreamento Intensificado para Indivíduos de Alto Risco
Indivíduos identificados com risco elevado para câncer hereditário, como portadores de mutações patogênicas germinativas ou aqueles com um histórico familiar fortemente sugestivo, requerem protocolos de vigilância e rastreamento diferenciados e mais intensivos em comparação com as diretrizes populacionais gerais. O objetivo primário é a detecção precoce das neoplasias, visando melhorar o prognóstico.
Rastreamento de Câncer de Mama
Para mulheres com risco significativamente aumentado, como as portadoras de mutações nos genes BRCA1/2 ou com outros fatores de alto risco definidos, o protocolo de rastreamento mamário é intensificado. Geralmente, recomenda-se a combinação de mamografia anual com ressonância magnética (RM) das mamas anual, frequentemente realizadas de forma intercalada a cada seis meses. O início deste rastreamento é antecipado, usualmente entre 25 e 30 anos de idade, ajustado conforme a mutação específica e o histórico familiar. Adicionalmente, exames clínicos das mamas são recomendados em intervalos mais curtos, a cada 6 a 12 meses.
Rastreamento de Câncer de Ovário
O rastreamento para câncer de ovário é uma consideração importante para mulheres com risco elevado, particularmente portadoras de mutações BRCA1/2. As estratégias atuais podem envolver a realização de ultrassonografia transvaginal e a dosagem sérica do marcador CA-125. Contudo, é crucial reconhecer as limitações significativas e a baixa eficácia comprovada destes métodos para a detecção precoce e confiável do câncer de ovário em estágio inicial. Dada esta dificuldade no rastreamento efetivo, a discussão sobre estratégias de redução de risco, como a salpingooforectomia bilateral redutora de risco (RRSO), torna-se um componente central do manejo após a conclusão da prole. A vigilância para portadoras de mutações BRCA também pode incluir exames ginecológicos regulares.
Rastreamento de Outras Neoplasias Associadas
A vigilância deve ser estendida a outros cânceres associados a síndromes hereditárias específicas, com estratégias individualizadas:
- Câncer de Próstata: Homens portadores de mutações BRCA1/2, sobretudo BRCA2, possuem risco aumentado de câncer de próstata, incluindo formas agressivas e de início precoce. O rastreamento é individualizado com base na mutação, idade e histórico familiar, podendo envolver dosagem de PSA e exame retal digital em idade mais jovem do que o recomendado para a população geral.
- Câncer de Pâncreas: Portadores de mutações BRCA1/2 e de outras síndromes (como Lynch e Peutz-Jeghers) têm risco elevado. O screening para câncer de pâncreas é complexo e geralmente considerado em contextos de pesquisa ou para indivíduos com histórico familiar muito forte, podendo envolver exames de imagem como ultrassonografia endoscópica ou RM/colangiopancreatografia por RM (CPRM), mas não é rotineiramente recomendado para todos os portadores de mutação.
- Câncer Gástrico (associado ao CDH1): Para portadores de mutação no gene CDH1, diante do risco extremamente elevado de câncer gástrico difuso, a vigilância endoscópica pode ser considerada. No entanto, a gastrectomia total profilática é frequentemente a principal estratégia de manejo de risco discutida.
- Câncer Colorretal (Síndrome de Lynch): Para indivíduos diagnosticados com Síndrome de Lynch, a colonoscopia intensiva é fundamental. Recomenda-se a realização de colonoscopias a cada 1-2 anos, iniciando em idade jovem (geralmente entre 20-25 anos ou antes, dependendo do histórico familiar).
A definição da estratégia de rastreamento para indivíduos de alto risco deve ser sempre personalizada, resultante de aconselhamento genético detalhado, avaliação por equipe multidisciplinar e considerando as preferências informadas do paciente.
Opções de Redução de Risco: Quimioprevenção Farmacológica
A quimioprevenção farmacológica representa uma estratégia de redução de risco de câncer de mama para indivíduos selecionados de alto risco, utilizando medicamentos específicos para diminuir a probabilidade de desenvolvimento da neoplasia mamária. As principais classes de agentes utilizadas são os Moduladores Seletivos do Receptor de Estrogênio (SERMs) e os Inibidores da Aromatase (IAs).
Moduladores Seletivos do Receptor de Estrogênio (SERMs)
Os SERMs, como o tamoxifeno e o raloxifeno, atuam bloqueando seletivamente os receptores de estrogênio no tecido mamário. Estudos demonstram que seu uso pode reduzir a incidência de câncer de mama com receptores de estrogênio (RE) positivos em aproximadamente 50% nas populações de alto risco. A indicação, no entanto, requer uma avaliação cuidadosa dos riscos associados, que incluem um aumento na incidência de eventos tromboembólicos e, especificamente para o tamoxifeno, um risco elevado de câncer de endométrio.
Inibidores da Aromatase (IAs)
Os Inibidores da Aromatase são outra opção farmacológica, particularmente indicados para mulheres na pós-menopausa. Eles agem inibindo a enzima aromatase, responsável pela conversão periférica de androgênios em estrogênios, diminuindo assim os níveis circulantes deste hormônio. Similarmente aos SERMs, os IAs demonstraram reduzir o risco de câncer de mama RE-positivo em cerca de 50%. Os efeitos adversos potenciais incluem um risco aumentado de osteoporose, fraturas e sintomas musculoesqueléticos.
Considerações na Indicação e Individualização
A aplicabilidade da quimioprevenção em portadores de mutações BRCA1/2 necessita de análise criteriosa. Dado que os SERMs e IAs atuam primariamente na prevenção de tumores RE-positivos, sua eficácia pode ser limitada em portadoras de mutações BRCA1, frequentemente associadas a tumores triplo-negativos. Portadoras de mutações BRCA2, mais comumente associadas a tumores RE-positivos, podem ter maior benefício. É importante notar que o impacto destas medicações na redução do risco de câncer de ovário é limitado. A decisão pela quimioprevenção deve ser rigorosamente individualizada, considerando o perfil de risco específico (mutação, histórico), os benefícios potenciais diretos, os riscos e efeitos colaterais das medicações, as comorbidades da paciente e suas preferências pessoais, sempre no contexto de uma discussão detalhada e tomada de decisão compartilhada.
Opções de Redução de Risco: Cirurgias Profiláticas (Redutoras de Risco)
Para indivíduos identificados com mutações genéticas que conferem um risco substancialmente elevado para o desenvolvimento de câncer, como portadores de mutações em BRCA1/2, TP53, PTEN ou CDH1, as intervenções cirúrgicas profiláticas representam a estratégia mais eficaz disponível para a redução desse risco. A decisão pela realização de tais procedimentos deve ser sempre individualizada, precedida por aconselhamento genético detalhado e avaliação multidisciplinar, ponderando os riscos e benefícios, as preferências do paciente e o impacto potencial na qualidade de vida.
Mastectomia Bilateral Redutora de Risco (MRR)
A Mastectomia Redutora de Risco (MRR), também conhecida como adeno-mastectomia bilateral profilática, é uma intervenção cirúrgica primária para mulheres com risco significativamente aumentado de câncer de mama (ex: portadoras de mutações BRCA1/2, TP53, PTEN). A abordagem cirúrgica frequentemente visa a preservação da pele e, quando oncológicamente seguro, do complexo areolopapilar, permitindo opções de reconstrução mamária imediata ou tardia para otimizar os resultados estéticos e psicológicos.
- Eficácia e Limitações: A MRR demonstra uma eficácia notável, reduzindo o risco de desenvolvimento de câncer de mama em aproximadamente 90%. No entanto, é fundamental compreender que a redução de risco não é absoluta. Devido à possibilidade de permanência de tecido mamário residual mínimo, um risco, embora muito baixo, de desenvolvimento posterior da neoplasia persiste.
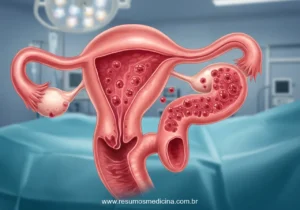
Salpingo-ooforectomia Bilateral Redutora de Risco (RRSO)
A RRSO envolve a remoção cirúrgica de ambos os ovários e ambas as trompas de Falópio. É uma recomendação forte para mulheres portadoras de mutações nos genes BRCA1/2, particularmente após a conclusão da prole e idealmente antes da menopausa fisiológica (geralmente indicada entre 35 e 45 anos, ajustada pela mutação específica e histórico familiar).
- Impacto na Redução de Risco: A RRSO reduz drasticamente o risco de câncer de ovário e de trompas de Falópio (atualmente reconhecidas como sítio de origem frequente para cânceres serosos de alto grau). Adicionalmente, confere uma redução secundária no risco de câncer de mama, especialmente quando realizada no período pré-menopausa, devido à cessação da produção ovariana de estrogênio.
- Consequências e Limitações: A principal consequência da RRSO em mulheres na pré-menopausa é a indução de menopausa cirúrgica imediata. O manejo dos sintomas associados (vasomotores, urogenitais) e a prevenção de complicações a longo prazo (saúde óssea, cardiovascular) requerem avaliação cuidadosa, incluindo a discussão sobre terapia hormonal, se aplicável. Importante ressaltar que a RRSO não elimina o risco de desenvolvimento de câncer peritoneal primário, uma neoplasia histologicamente similar ao câncer epitelial ovariano.
Gastrectomia Total Profilática
Para indivíduos portadores de mutações germinativas patogênicas no gene CDH1, associadas a um risco cumulativo muito elevado de Câncer Gástrico Difuso Hereditário (CGDH), a gastrectomia total profilática é uma consideração importante. Dado o padrão infiltrativo e o diagnóstico frequentemente tardio do CGDH (tipo difuso, com células em anel de sinete, conforme classificação de Lauren), esta cirurgia preventiva é discutida como a principal estratégia de redução de risco após aconselhamento genético e avaliação multidisciplinar.
Conclusão: Integrando a Genética no Manejo Oncológico Personalizado
A incorporação do conhecimento sobre hereditariedade no câncer representa um avanço fundamental na oncologia moderna. A identificação de síndromes de predisposição genética – exemplificadas pelas mutações em genes como BRCA1, BRCA2, TP53, PTEN, CDH1 e os genes de reparo de mismatch associados à Síndrome de Lynch – é imperativa, dado o risco substancialmente elevado e a frequente ocorrência de neoplasias em idades mais jovens associadas a essas condições.
O reconhecimento de indivíduos e famílias sob risco aumentado fundamenta-se na avaliação criteriosa do histórico familiar e pessoal, sendo o aconselhamento genético especializado um passo subsequente indispensável. Este processo facilita a tomada de decisão informada sobre a pertinência e as implicações da testagem genética, orientada por critérios clínicos estabelecidos.
A confirmação de uma mutação germinativa patogênica direciona para um manejo clínico multidisciplinar e intensamente personalizado. Este manejo engloba estratégias de vigilância intensificada, adaptadas ao perfil de risco específico (como a combinação de mamografia e ressonância magnética para portadoras de mutações BRCA), e a consideração de intervenções de redução de risco. Tais intervenções incluem a quimioprevenção farmacológica e as cirurgias profiláticas (mastectomia, salpingo-ooforectomia, ou gastrectomia, conforme a síndrome). A decisão final sobre a estratégia a ser adotada deve ser individualizada, resultante de uma discussão aprofundada que pondere o risco quantificado, as comorbidades, e os valores e preferências do paciente.
A integração da informação genética no cuidado oncológico permanece em franca evolução. As perspectivas futuras apontam para a contínua descoberta de genes de suscetibilidade, um entendimento mais profundo dos fatores que modulam a penetrância das mutações (incluindo interações gene-ambiente) e o desenvolvimento de terapias cada vez mais refinadas. Estas avanços consolidam a transição para uma oncologia de precisão que considera não apenas as características moleculares do tumor, mas também a constituição genética germinativa do indivíduo.